Kapitel 1. Wie man Bioinformatik lernt
Diese Arbeit wurde mithilfe von KI übersetzt. Wir freuen uns über dein Feedback und deine Kommentare: translation-feedback@oreilly.com
In den Laboren auf der ganzen Welt sequenzieren Maschinen gerade die Genome des Lebens auf der Erde. Trotz rapide sinkender Kosten und enormer technologischer Fortschritte bei der Genomsequenzierung sehen wir nur einen kleinen Ausschnitt der biologischen Informationen, die in jeder Zelle, jedem Gewebe, jedem Organismus und jedem Ökosystem enthalten sind. Doch der Bruchteil der gesamten biologischen Informationen, die wir sammeln, bedeutet Berge von Daten, mit denen Biologen arbeiten müssen. Zu keinem anderen Zeitpunkt in der Geschichte der Menschheit war unsere Fähigkeit, die Komplexität des Lebens zu verstehen, so sehr von unserer Fähigkeit abhängig, mit Daten zu arbeiten und sie zu analysieren.
In diesem Buch geht es darum, Bioinformatik durch die Entwicklung von Datenkompetenz zu lernen. In diesem Kapitel erfahren wir, was Datenkompetenz ist und warum das Erlernen von Datenkompetenz der beste Weg ist, Bioinformatik zu lernen. Außerdem werden wir uns ansehen, was robuste und reproduzierbare Forschung ausmacht.
Warum Bioinformatik? Die wachsende Datenmenge der Biologie
Bioinformatikerinnen und Bioinformatiker befassen sich mit der Gewinnung biologischer Erkenntnisse aus großen Datenmengen mit speziellen Fähigkeiten und Werkzeugen. In der Frühzeit der Biologie waren die Datenmengen klein und überschaubar. Die meisten Biologinnen und Biologen konnten ihre Daten selbst analysieren, nachdem sie einen Statistikkurs besucht hatten, und zwar mit Microsoft Excel auf einem Desktop-Computer. Doch das ändert sich jetzt rapide. Große Sequenzierungsdatensätze sind weit verbreitet und werden in Zukunft noch weiter verbreitet sein. Um diese Daten zu analysieren, braucht man andere Werkzeuge, neue Fähigkeiten und viele Computer mit viel Speicher, Rechenleistung und Festplattenplatz.
In relativ kurzer Zeit sind die Kosten für die Sequenzierung drastisch gesunken, so dass Forscher die Sequenzierungsdaten nutzen können, um wichtige biologische Fragen zu beantworten. Früher war die Sequenzierung mit geringem Durchsatz verbunden und kostspielig. Die Sequenzierung ganzer Genome war teuer (das menschliche Genom kostete etwa 2,7 Milliarden Dollar) und nur durch große Kooperationen möglich. Seit der Veröffentlichung des menschlichen Genoms sind die Kosten für die Sequenzierung bis etwa 2008 exponentiell gesunken, wie in Abbildung 1-1 dargestellt. Mit der Einführung der Sequenzierungstechnologien der nächsten Generation fielen die Kosten für die Sequenzierung einer Megabase DNA noch schneller. An diesem entscheidenden Punkt wurde eine Technologie, die bisher nur für große Sequenzierungsprojekte (oder einzelne Forscher/innen mit sehr tiefen Taschen) erschwinglich war, für Forscher/innen aus der gesamten Biologie erschwinglich. Wahrscheinlich liest du dieses Buch, um zu lernen, mit Sequenzierungsdaten zu arbeiten, deren Erstellung vor weniger als 10 Jahren noch viel zu teuer gewesen wäre.
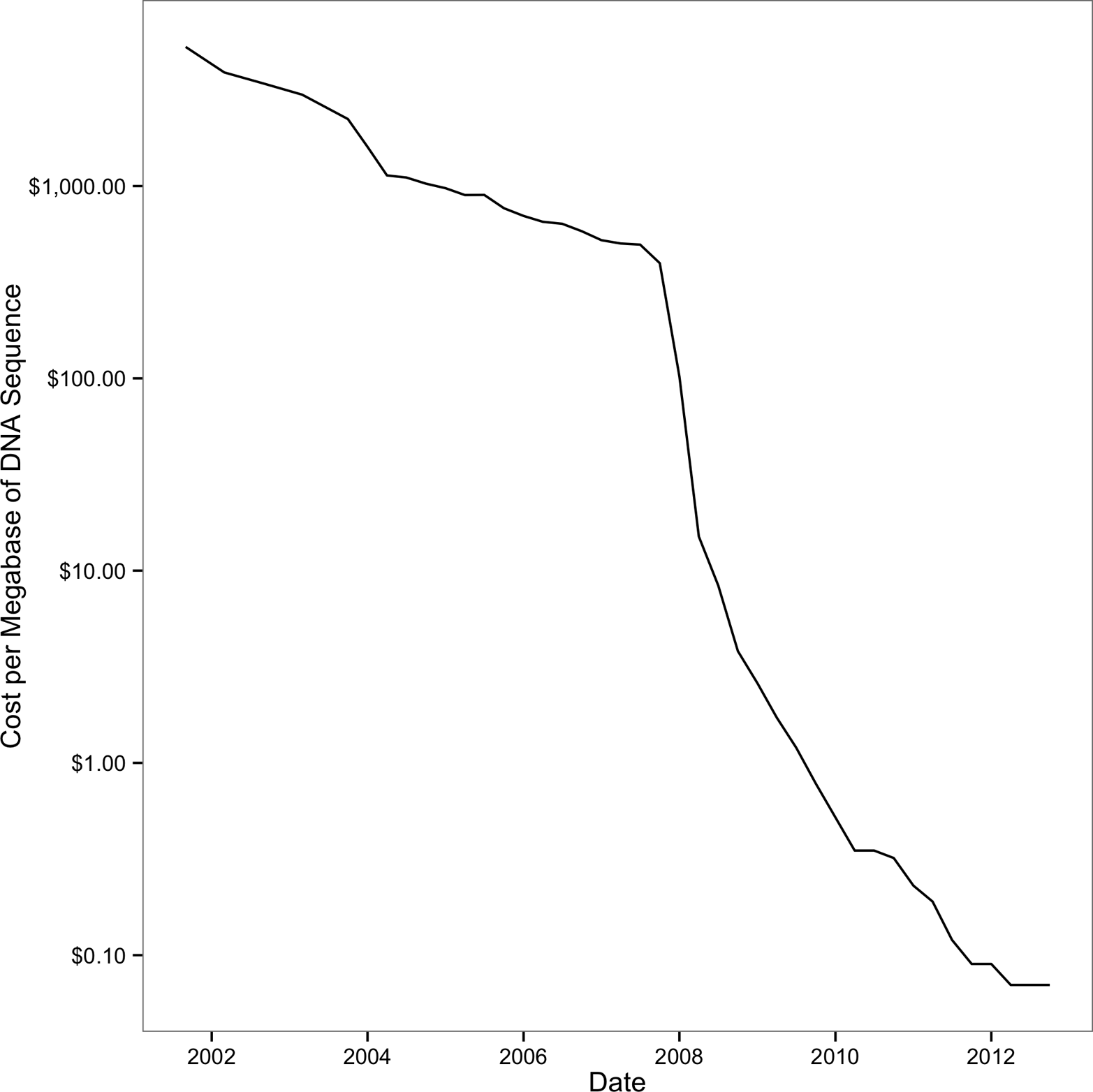
Abbildung 1-1. Rückgang der Sequenzierungskosten (beachte, dass die y-Achse eine logarithmische Skala hat); der starke Rückgang um 2008 war auf die Einführung der Sequenzierungsdaten der nächsten Generation zurückzuführen. (Abbildung reproduziert und Daten heruntergeladen von den NIH)
Was war die Folge des Rückgangs der Sequenzierungskosten aufgrund dieser neuen Technologien? Wie du vielleicht schon vermutet hast, viele, viele Daten. Biologische Datenbanken sind nach einem exponentiellen Wachstum mit Daten überschwemmt worden. Während früher kleine Datenbanken, die von Mitarbeitern gemeinsam genutzt wurden, ausreichten, liegen heute Petabytes an nützlichen Daten auf Servern auf der ganzen Welt. Wichtige Erkenntnisse über biologische Fragen sind nicht nur in den unanalysierten Versuchsdaten auf deiner Festplatte gespeichert, sondern auch auf einer Festplatte in einem Rechenzentrum, das Tausende von Kilometern entfernt ist.
Das Wachstum der biologischen Datenbanken ist ebenso erstaunlich wie der Rückgang der Sequenzierungskosten. Ein Beispiel dafür ist das Sequence Read Archive (früher bekannt als Short Read Archive), ein Speicher für die Rohdaten von Sequenzierungsexperimenten. Seit 2010 hat es ein bemerkenswertes Wachstum erlebt; siehe Abbildung 1-2.
Um dieses unglaubliche Wachstum der Sequenzierungsdaten in einen Zusammenhang zu bringen, betrachte das Mooresche Gesetz. Gordon Moore (ein Mitbegründer von Intel) stellte fest, dass sich die Anzahl der Transistoren in Computerchips etwa alle zwei Jahre verdoppelt. Mehr Transistoren pro Chip bedeuten höhere Geschwindigkeiten bei Computerprozessoren und mehr Arbeitsspeicher in Computern, was zu leistungsfähigeren Computern führt. Dieses außergewöhnliche Tempo der technologischen Verbesserung - eine Verdopplung der Leistung alle zwei Jahre - ist wahrscheinlich das schnellste technologische Wachstum, das die Menschheit je erlebt hat. Doch seit 2011 hat die Menge der im Short Read Archive gespeicherten Sequenzierungsdaten sogar dieses unglaubliche Wachstum übertroffen und sich jedes Jahr verdoppelt.
Um die Sache noch komplizierter zu machen, werden ständig neue Tools für die Analyse biologischer Daten entwickelt, und die zugrunde liegenden Algorithmen werden weiterentwickelt. In einem Bericht aus dem Jahr 2012 wurden über 70 Short-Read-Mapper aufgelistet (Fonseca et al., 2012; siehehttp://bit.ly/hts-mappers). Auch unsere Herangehensweise an die Genomassemblierung hat sich in den letzten fünf Jahren stark verändert, da Methoden zur Assemblierung langer Sequenzen (wie z. B. Overlap-Layout-Consensus-Algorithmen) mit dem Aufkommen kurzer High-Throughput-Sequencing-Reads aufgegeben wurden. Jetzt führen die Fortschritte in der Sequenzierungschemie zu längeren Sequenzierungslängen und neue Algorithmen ersetzen andere, die erst wenige Jahre alt waren.
Leider hat diese Fülle und schnelle Entwicklung von Bioinformatik-Tools auch ihre Schattenseiten. Oft werden Bioinformatik-Tools nicht angemessen verglichen, oder wenn doch, dann nur mit einem Organismus. Das macht es für neue Biologen schwierig, das beste Tool für die Analyse ihrer Daten zu finden und auszuwählen. Erschwerend kommt hinzu, dass einige Bioinformatikprogramme nicht aktiv weiterentwickelt werden, so dass sie an Relevanz verlieren oder Fehler enthalten, die die Ergebnisse negativ beeinflussen können. All das macht die Wahl eines geeigneten Bioinformatikprogramms für deine eigene Forschung schwierig. Noch wichtiger ist aber, dass du die Ergebnisse von Bioinformatikprogrammen, die mit deinen eigenen Daten arbeiten, kritisch bewertest. In Teil II werden wir Beispiele dafür sehen, wie Datenkenntnisse uns helfen können, die Ergebnisse von Programmen zu bewerten.
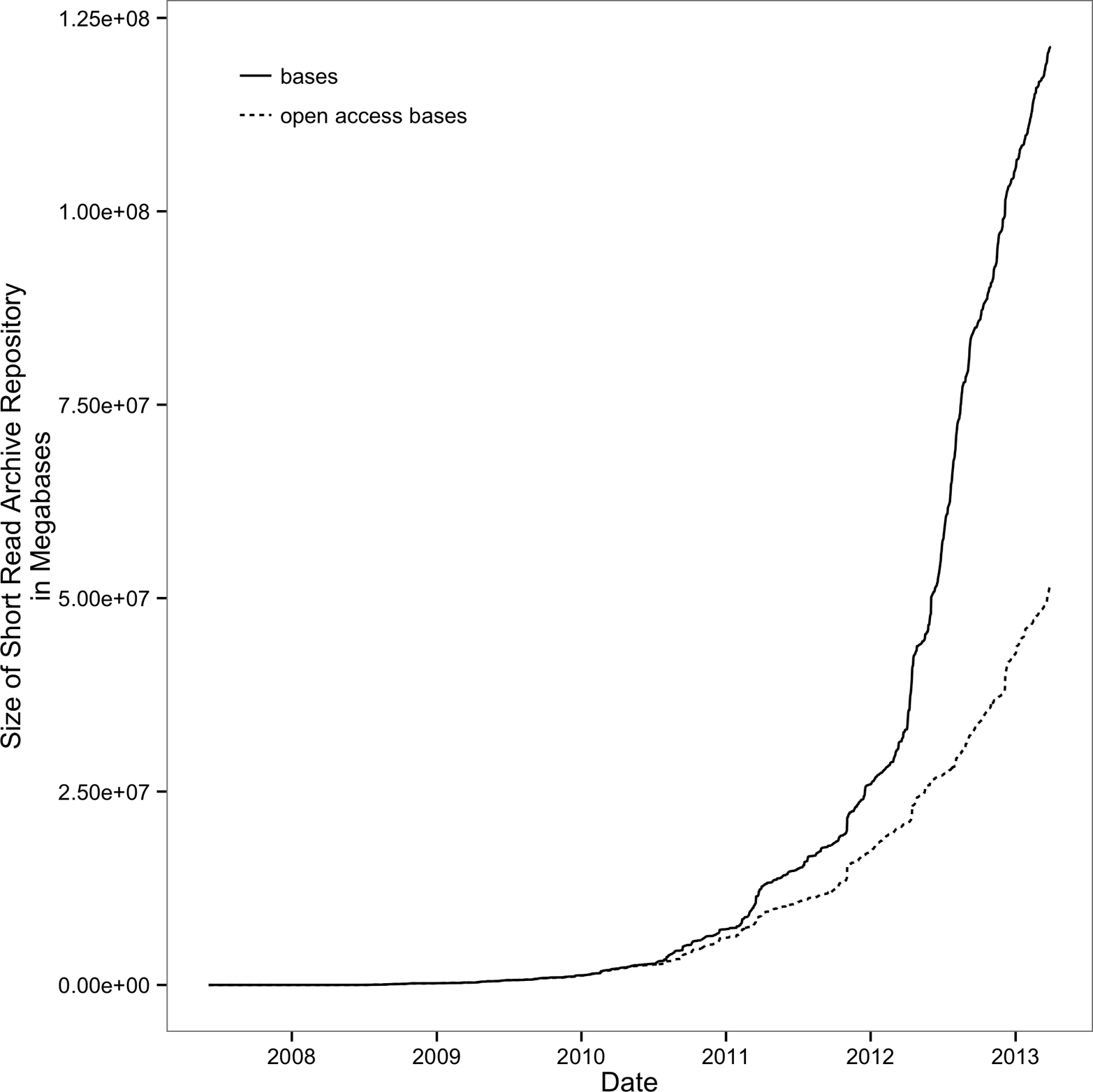
Abbildung 1-2. Exponentielles Wachstum des Short Read Archive; Open-Access-Basen sind öffentlich zugängliche SRA-Beiträge (Abbildung reproduziert und Daten heruntergeladen von den NIH)
Datenkompetenz lernen, um Bioinformatik zu lernen
Da sich die Art der biologischen Daten so schnell verändert, wie soll man da Bioinformatik lernen? Wie soll eine Biologin oder ein Biologe bei all den Tools, die es gibt und die ständig weiterentwickelt werden, wissen, ob ein Programm für die Daten ihres oder seines Organismus geeignet ist?
Die Lösung ist, an die Bioinformatik heranzugehen, wie es ein Bioinformatiker tut: etwas ausprobieren und die Ergebnisse auswerten. Auf diese Weise geht es in der Bioinformatik nur darum, mit Daten am Computer zu experimentieren und die Ergebnisse zu verstehen. Der experimentelle Teil ist einfach; das ist für die meisten Wissenschaftler/innen selbstverständlich. Der begrenzende Faktor für die meisten Biologen ist die Fähigkeit, mit Daten zu experimentieren und mit großen Datenmengen am Computer zu arbeiten. Das Ziel dieses Buches ist es, dir die notwendigen bioinformatischen Datenkenntnisse zu vermitteln, damit du mit Daten am Computer genauso einfach experimentieren kannst wie im Labor.
Leider sind viele der gängigen Berechnungswerkzeuge von Biologen nicht für die Größe und Komplexität moderner biologischer Daten geeignet. Komplexe Datenformate, die Verknüpfung zahlreicher Programme und die Bewertung von Software und Daten machen die Arbeit mit großen bioinformatischen Datensätzen schwierig. Das Erlernen von Grundkenntnissen in der Bioinformatik gibt dir die Grundlage, um jedes Bioinformatikprogramm und jede Analysemethode zu erlernen, anzuwenden und zu bewerten. In 10 Jahren werden Bioinformatiker/innen vielleicht nur noch einige wenige der heute verfügbaren Bioinformatik-Programme benutzen. Aber wir werden mit Sicherheit Datenkenntnisse und Experimente nutzen, um Daten und Methoden der Zukunft zu bewerten.
Was sind also Datenkompetenzen? Es handelt sich dabei um eine Reihe von Computerkenntnissen, die es dir ermöglichen, komplexe Datensätze mit Hilfe von bekannten Werkzeugen schnell und improvisiert zu betrachten. Eine gute Analogie ist das, was Jazzmusiker als "Chops" bezeichnen. Ein Jazzmusiker mit guten Fähigkeiten kann in einen Nachtclub gehen, einen bekannten Standardsong hören, die Akkordwechsel erkennen und anfangen, musikalische Ideen über diese Akkorde zu spielen. Genauso kann ein Bioinformatiker mit guten Datenkenntnissen einen riesigen Sequenzierungsdatensatz erhalten und sofort damit beginnen, eine Reihe von Werkzeugen zu benutzen, um zu sehen, welche Geschichte die Daten erzählen.
Wie ein Jazzmusiker, der sein Instrument beherrscht, beherrscht auch ein Bioinformatiker mit ausgezeichneten Datenkenntnissen eine Reihe von Werkzeugen. Das Erlernen der Werkzeuge ist ein notwendiger, aber nicht ausreichender Schritt, um Datenkenntnisse zu entwickeln (ähnlich wie das Erlernen eines Instruments ein notwendiger, aber nicht ausreichender Schritt ist, um musikalische Ideen zu spielen). Im Laufe des Buches werden wir unsere Datenkenntnisse weiterentwickeln, angefangen beim Einrichten eines Bioinformatikprojekts und der Daten in Teil II bis hin zum Erlernen kleiner und großer Tools für die Datenanalyse in Teil III. Dieses Buch kann dich jedoch nur auf den richtigen Weg bringen; wirkliche Meisterschaft erfordert das Lernen durch die wiederholte Anwendung von Fähigkeiten auf reale Probleme.
Neue Herausforderungen für reproduzierbare und robuste Forschung
Die zunehmende Nutzung großer Sequenzierdatensätze in der Biologie verändert nicht nur die Werkzeuge und Fähigkeiten, die wir brauchen: Sie verändert auch die Reproduzierbarkeit und Robustheit unserer wissenschaftlichen Ergebnisse. Während wir neue Werkzeuge und Fähigkeiten zur Analyse von Genomikdaten einsetzen, müssen wir sicherstellen, dass unsere Ansätze genauso reproduzierbar und robust sind wie alle anderen experimentellen Ansätze. Leider machen der Umfang unserer Daten und die Komplexität unserer Analyse-Workflows dieses Ziel in der Genomik besonders schwierig.
Die Voraussetzung für Reproduzierbarkeit ist dass wir unsere Daten und Methoden teilen. In der Zeit vor der Genomik war das viel einfacher. Veröffentlichungen konnten detaillierte Zusammenfassungen der Methoden und ganze Datensätze enthalten - genau wie Kreitmans Veröffentlichung von 1986 mit einer 4.713bp flankierenden Sequenz des Adh-Gens (sie war in der Mitte der Veröffentlichung eingebettet). Heute haben die Artikel lange zusätzliche Methoden, Codes und Daten. Auch die gemeinsame Nutzung von Daten ist nicht mehr trivial, denn Sequenzierungsprojekte können Terabytes an begleitenden Daten enthalten. Referenzgenome und Annotationsdatensätze, die in Analysen verwendet werden, werden ständig aktualisiert, was die Reproduzierbarkeit erschweren kann. Links zu ergänzenden Materialien, Methoden und Daten auf Zeitschriften-Websites gehen kaputt, Materialien auf Fakultäts-Websites verschwinden, wenn Fakultätsmitglieder umziehen oder ihre Websites aktualisieren, und Softwareprojekte veralten, wenn die Entwickler ausscheiden und den Code nicht aktualisieren. Im Laufe dieses Buches werden wir uns ansehen, was du tun kannst, um die Reproduzierbarkeit deines Projekts zu verbessern, während du die eigentliche Analyse durchführst, denn ich bin der Meinung, dass diese beiden Aktivitäten sich gegenseitig ergänzen müssen.
Außerdem kann die Komplexität der bioinformatischen Analysen dazu führen, dass die Ergebnisse anfällig für Fehler und technische Verwechslungen sind. Selbst bei relativ routinemäßigen Genomprojekten können Dutzende verschiedener Programme, komplizierte Kombinationen von Eingabeparametern und viele Proben- und Annotationsdatensätze zum Einsatz kommen; außerdem kann die Arbeit auf verschiedene Server und Workstations verteilt sein. All diese Datenverarbeitungsschritte führen zu Ergebnissen, die in übergeordneten Analysen verwendet werden, aus denen wir unsere biologischen Schlussfolgerungen ziehen. Das Endergebnis ist, dass die Forschungsergebnisse auf einem wackeligen Gerüst aus zahlreichen Verarbeitungsschritten beruhen können. Erschwerend kommt hinzu, dass Bioinformatik-Workflows und -Analysen in der Regel nur einmal ausgeführt werden, um Ergebnisse für eine Veröffentlichung zu erzielen, und dann nie wieder ausgeführt oder getestet werden. Diese Analysen können von sehr spezifischen Versionen der verwendeten Software abhängen, was die Reproduktion auf einem anderen System schwierig machen kann. Beim Erlernen bioinformatischer Datenkenntnisse ist es notwendig, gleichzeitig die Reproduzierbarkeit und bewährte Methoden zu erlernen. Werfen wir einen Blick auf Reproduzierbarkeit und Robustheit.
Reproduzierbare Forschung
Die Reproduktion wissenschaftlicher Erkenntnisse ist der einzige Weg, um zu bestätigen, dass sie korrekt sind und nicht das Artefakt eines einzelnen Experiments oder einer Analyse. Karl Popper sagte in The Logic of Scientific Discovery (Die Logik der wissenschaftlichen Entdeckung): "Nicht reproduzierbare Einzelereignisse sind für die Wissenschaft ohne Bedeutung" (1959). Die unabhängige Replikation von Experimenten und Analysen ist der Goldstandard, nach dem wir die Gültigkeit wissenschaftlicher Erkenntnisse beurteilen. Leider sind die meisten Sequenzierungsexperimente zu teuer, um sie vom Reagenzglas aufwärts zu reproduzieren, so dass wir uns zunehmend nur auf die In-silico-Reproduzierbarkeit verlassen. Die Komplexität von Bioinformatikprojekten schreckt in der Regel von der Reproduktion ab. Deshalb ist es unsere Aufgabe als gute Wissenschaftler/innen, die In-silico-Reproduzierbarkeit zu erleichtern und zu fördern, indem wir sie vereinfachen. Wie wir später sehen werden, kann die Anwendung guter Reproduzierbarkeitspraktiken auch dein Leben als Forscher/in einfacher machen.
Was ist also ein reproduzierbares Bioinformatikprojekt? Zumindest bedeutet es, dass du den Code und die Daten deines Projekts weitergibst. Die meisten Zeitschriften und Förderorganisationen verlangen von dir, dass du die Daten deines Projekts weitergibst, und zu diesem Zweck gibt es Ressourcen wie das Sequence Read Archive des NCBI. Redakteure und Gutachter werden oft vorschlagen (oder in manchen Fällen verlangen), dass auch der Code eines Projekts geteilt wird, vor allem, wenn der Code ein wichtiger Teil der Ergebnisse einer Studie ist. Aber wir können und sollten noch viel mehr tun, um die Reproduzierbarkeit unserer Projekte zu gewährleisten. Durch die Reproduktion von bioinformatischen Analysen zur Überprüfung der Ergebnisse habe ich gelernt, dass der Teufel im Detail steckt.
Zum Beispiel hatten meine Kollegen und ich einmal Schwierigkeiten, eine RNA-seq-Analyse der differentiellen Expression zu reproduzieren, die wir selbst durchgeführt hatten. Wir hatten vorläufige Ergebnisse aus einer Analyse einer Untergruppe von Proben, die wir einige Wochen zuvor durchgeführt hatten, aber zu unserer Überraschung ergab unsere aktuelle Analyse eine drastisch kleinere Gruppe von unterschiedlich exprimierten Genen. Nachdem wir noch einmal überprüft hatten, wie unsere früheren Ergebnisse entstanden waren, die Datenversionen und die Erstellungszeiten der Dateien verglichen und die Unterschiede im Analysecode untersucht hatten, waren wir immer noch ratlos - nichts konnte den Unterschied zwischen den Ergebnissen erklären. Schließlich überprüften wir die Version unseres R-Pakets und stellten fest, dass es auf unserem Server aktualisiert worden war. Wir installierten die alte Version erneut, um sicherzugehen, dass dies die Ursache für den Unterschied war - und tatsächlich war es so. Die Lektion hier ist, dass die Replikation, entweder durch dich in der Zukunft oder durch jemand anderen, oft nicht nur von Daten und Code abhängt, sondern auch von Details wie Softwareversionen und wann die Daten heruntergeladen wurden und welche Version sie haben. Diese Metadaten, also Daten über Daten, sind ein entscheidendes Detail, um die Reproduzierbarkeit sicherzustellen.
Eine weitere motivierende Fallstudie zur Reproduzierbarkeit in der Bioinformatik ist die sogenannte "Duke Saga". Dr. Anil Potti und andere Forscher an der Duke University entwickelten eine Methode, mit der Expressionsdaten aus Hochdurchsatz-Mikroarrays verwendet wurden, um das Ansprechen auf verschiedene Chemotherapeutika zu erkennen und vorherzusagen. Diese Methoden waren der Beginn einer neuen Stufe der personalisierten Medizin und wurden eingesetzt, um die Chemotherapie-Behandlungen für Patienten in klinischen Studien zu bestimmen. Zwei Biostatistiker, Keith Baggerly und Kevin Coombes, fanden jedoch bei dem Versuch, diese Studie zu reproduzieren, schwerwiegende Fehler in der Analyse (Baggerly und Coombes, 2009). Viele davon erforderten das, was Baggerly und Coombes als "forensische Bioinformatik" bezeichneten - Detektivarbeit, um zu versuchen, die Ergebnisse einer Studie zu reproduzieren, wenn es keine ausreichende Dokumentation gibt, um jeden Schritt nachzuvollziehen. Insgesamt fanden Baggerly und Coombes mehrere schwerwiegende Fehler, darunter:
-
Ein Off-by-One-Fehler, da eine ganze Liste von Genexpressionswerten im Verhältnis zu ihrem korrekten Bezeichner nach unten verschoben wurde
-
Zwei Ausreißergene von biologischem Interesse waren nicht auf den verwendeten Microarrays
-
Es gab eine Verwechslung der Behandlung mit dem Tag, an dem das Microarray durchgeführt wurde
-
Die Namen der Mustergruppen wurden verwechselt
Die Arbeit von Baggerly und Coombes lässt sich am besten in ihrem Open-Access-Artikel "Deriving Chemosensitivity from Cell Lines" zusammenfassen: Forensic Bioinformatics and Reproducible Research in High-Throughput Biology" (siehe das GitHub-Verzeichnis dieses Kapitels für diesen Artikel und weitere Informationen über die Duke Saga). Die Lehre aus der Arbeit von Baggerly und Coombes lautet: "Häufige Fehler sind einfach, und einfache Fehler sind häufig", und eine schlechte Dokumentation kann sowohl zu Fehlern als auch zu Irreproduzierbarkeit führen. Eine Dokumentation der Methoden, Datenversionen und des Codes hätte nicht nur die Reproduzierbarkeit erleichtert, sondern wahrscheinlich auch einige der schweren Fehler in ihrer Studie verhindert. Wenn du dich um maximale Reproduzierbarkeit in deinem Projekt bemühst, wird deine Arbeit auch robuster.
Robuste Forschung und die goldene Regel der Bioinformatik
Da der Computer ein so scharfes Werkzeug ist, dass er wirklich nützlich ist, kannst du dich an ihm schneiden .
Die technischen Fertigkeiten der Statistik (1964)John Tukey
Wenn in der Biologie im Nasslabor Experimente fehlschlagen, kann das sehr offensichtlich sein, aber in der Informatik ist das nicht immer der Fall. Elektrophorese-Gele, die eher wie Rorschach-Flecken als wie ordentliche Banden aussehen, sind ein deutliches Zeichen dafür, dass etwas schief gelaufen ist. Beim wissenschaftlichen Rechnen können Fehler stillschweigend auftreten, d. h. Code und Programme können eine Ausgabe produzieren (anstatt mit einem Fehler aufzuhören), aber diese Ausgabe kann falsch sein. Das ist ein sehr wichtiger Gedanke, den du beim Lernen der Bioinformatik im Hinterkopf behalten solltest.
Außerdem ist es in der Wissenschaft üblich, dass der Code nur einmal ausgeführt wird, wenn die Forscher/innen das gewünschte Ergebnis erhalten und zum nächsten Schritt übergehen. Im Gegensatz dazu wird ein Videospiel auf Tausenden (wenn nicht gar Millionen) verschiedenen Rechnern ausgeführt und ständig von vielen Nutzern getestet. Wenn ein Fehler auftritt, der den Spielstand eines Nutzers löscht, ist es sehr wahrscheinlich, dass er schnell bemerkt und von den Nutzern gemeldet wird. Leider ist das bei den meisten Bioinformatikprojekten nicht der Fall.
Genomikdaten stellen auch eigene Herausforderungen für eine robuste Forschung dar. Erstens erzeugen die meisten Bioinformatik-Analysen Zwischenergebnisse, die zu groß und hochdimensional sind, um sie zu untersuchen oder einfach zu visualisieren. Selbst wenn es also möglich wäre, den gesamten Zwischendatensatz auf Probleme zu untersuchen, wäre es schwierig, dies für jeden einzelnen Schritt zu tun (daher greifen wir in der Regel darauf zurück, Stichproben der Daten zu untersuchen oder uns zusammenfassende Statistiken anzusehen). Zweitens ist es in der Biologie im Nasslabor meist einfacher, sich im Voraus ein Bild vom Ergebnis eines Experiments zu machen. So kann ein Forscher zum Beispiel erwarten, dass eine bestimmte mRNA in einigen Geweben in geringerer Menge exprimiert wird als ein bestimmtes Housekeeping-Gen. Mit diesen Erwartungen kann ein abweichendes Ergebnis auf einen fehlgeschlagenen Versuch und nicht auf die Biologie zurückgeführt werden. Im Gegensatz dazu macht es die hohe Dimensionalität der meisten Genomforschungsergebnisse fast unmöglich, eindeutige Vorab-Erwartungen zu bilden. Es ist unpraktisch, für jedes der Zehntausenden von Genen, die in einem RNA-seq-Experiment untersucht werden, spezifische Vorab-Erwartungen an die Expression zu stellen. Leider kann es ohne Vorab-Erwartungen ziemlich schwierig sein, gute von schlechten Ergebnissen zu unterscheiden.
Bioinformatiker müssen sich auch darüber im Klaren sein, dass Bioinformatik-Programme, selbst die großen, von der Community getesteten Tools wie Aligner und Assembler, bei ihrem speziellen Organismus möglicherweise nicht gut funktionieren. Organismen sind alle auf wundersame Weise verschieden, und ihre Genome sind es auch. Viele Bioinformatik-Tools wurden an einigen wenigen diploiden Modellorganismen wie dem Menschen getestet und weniger gut an den komplexen Genomen der anderen Teile des Lebensbaums. Erwarten wir wirklich, dass die Standardparameter eines Short-Read-Aligners, der auf menschliche Daten abgestimmt ist, bei einem polyploiden Genom funktionieren, das viermal so groß ist? Wahrscheinlich nicht.
Der einfachste Weg, um sicherzustellen, dass alles richtig funktioniert, ist, eine vorsichtige Haltung einzunehmen und alles zwischen den Berechnungsschritten zu überprüfen. Außerdem solltest du an biologische Daten (entweder aus einem Experiment oder aus einer Datenbank) mit einer gesunden Skepsis herangehen, dass etwas falsch sein könnte. In der Computerwelt ( ) ist dies mit dem Konzept "Garbage in, garbage out" verbunden - eine Analyse ist nur so gut wie die Daten, die hineingehen. In meinem Bioinformatikunterricht verwende ich diese Idee oft als Goldene Regel der Bioinformatik:
Das soll dich nicht paranoid machen, dass man keiner Bioinformatik trauen kann oder dass du jedes verfügbare Programm und jeden Parameter an deinen Daten testen musst. Vielmehr sollst du die gleiche vorsichtige Haltung einnehmen, die Softwareentwickler/innen und Bioinformatiker/innen auf die harte Tour gelernt haben. Die Überprüfung von Eingabedaten und Zwischenergebnissen, die schnelle Überprüfung der Korrektheit, die Einhaltung angemessener Kontrollen und das Testen von Programmen sind schon ein guter Anfang. Das bewahrt dich auch davor, dass du später auf Fehler stößt, deren Behebung bedeutet, dass du große Mengen an Arbeit wiederholen musst. Natürlich testest du, ob die Labortechniken funktionieren und konsistente Ergebnisse liefern.
Robuste und reproduzierbare Praktiken werden auch dein Leben einfacher machen
Die Arbeit in der Wissenschaft hat viele von uns einige Fakten des Lebens auf die harte Tour gelehrt. Diese sind wie Murphys Gesetz: Alles, was schiefgehen kann, wird es auch. Auch in der Bioinformatik gibt es solche Gesetze. Nachdem ich in diesem Bereich gearbeitet und Kriegsgeschichten mit anderen Bioinformatikern besprochen habe, kann ich praktisch garantieren, dass Folgendes passieren wird:
-
Mit ziemlicher Sicherheit wirst du eine Analyse mehr als einmal wiederholen müssen, möglicherweise mit neuen oder geänderten Daten. Häufig passiert das, weil du einen Fehler entdeckst, ein Mitarbeiter eine Datei hinzufügt oder aktualisiert oder du etwas Neues vor einem Schritt ausprobieren möchtest. In allen Fällen hängen nachgelagerte Analysen von diesen früheren Ergebnissen ab, was bedeutet, dass alle Schritte einer Analyse erneut durchgeführt werden müssen.
-
In der Zukunft wirst du (oder deine Mitarbeiter/innen oder dein/e Berater/in) mit ziemlicher Sicherheit einen Teil des Projekts wiederholen müssen und es wird völlig kryptisch aussehen. Dein einziger Schutz ist es, jeden Schritt zu dokumentieren. Wenn du die wichtigsten Fakten nicht aufschreibst (z. B. woher du die Daten heruntergeladen hast, wann du sie heruntergeladen hast und welche Schritte du durchgeführt hast), wirst du sie mit Sicherheit vergessen. Das Dokumentieren deiner Rechenarbeit entspricht dem Führen eines detaillierten Labornotizbuchs - ein absolut wichtiger Bestandteil der Wissenschaft.
Glücklicherweise hilft die Anwendung von Praktiken, die dein Projekt reproduzierbar machen, auch bei der Lösung dieser Probleme. In diesem Sinne machen gute Praktiken in der Bioinformatik (und im wissenschaftlichen Rechnen im Allgemeinen) das Leben einfacher und führen zu reproduzierbaren Projekten. Der Grund dafür ist einfach: Wenn jeder Schritt deines Projekts so konzipiert ist, dass er wiederholt werden kann (möglicherweise mit anderen Daten) und gut dokumentiert ist, ist es bereits auf dem besten Weg, reproduzierbar zu sein.
Wenn wir zum Beispiel Aufgaben mit einem Skript automatisieren und alle Eingabedaten und Softwareversionen aufzeichnen, können wir Analysen mit einem Tastendruck wiederholen. Die Reproduktion aller Schritte in diesem Skript ist viel einfacher, da ein gut geschriebenes Skript natürlich einen Arbeitsablauf dokumentiert (mehr dazu in Kapitel 12). Dieser Ansatz spart auch Zeit: Wenn du neue oder aktualisierte Daten erhältst, musst du das Skript nur mit der neuen Eingabedatei erneut ausführen. Das ist in der Praxis nicht schwer zu bewerkstelligen: Skripte sind nicht schwer zu schreiben und Computer sind hervorragend darin, sich wiederholende Aufgaben zu erledigen, die in einem Skript aufgezählt sind.
Empfehlungen für solide Forschung
Bei einer soliden Forschung geht es vor allem darum, eine Reihe von Praktiken anzuwenden, die die Chancen zu deinen Gunsten erhöhen , damit ein stiller Fehler deine Ergebnisse nicht verfälscht. Wie bereits erwähnt, sind die meisten dieser Praktiken auch aus anderen Gründen als der Vermeidung des gefürchteten stillen Fehlers von Vorteil - ein Grund mehr, die folgenden Empfehlungen bei deiner täglichen Arbeit in der Bioinformatik anzuwenden.
Achte auf die Versuchsplanung
Robuste Forschung beginnt mit einem guten Versuchsplan. Leider kann keine noch so brillante Analyse ein Experiment mit einem schlechten Design retten. Um einen brillanten Statistiker und Genetiker zu zitieren:
Den Statistiker nach dem Ende eines Experiments zu konsultieren, bedeutet oft nur, ihn zu bitten, eine Obduktion durchzuführen. Er kann vielleicht sagen, woran das Experiment gestorben ist.
R.A. Fischer
Dieses Zitat trifft den Nagel auf den Kopf: Ich habe schon Projekte gesehen, die auf meinem Schreibtisch gelandet sind, nachdem ich Tausende von Dollars für die Sequenzierung ausgegeben hatte, und die dann bei ihrer Ankunft völlig tot waren. Eine gute Versuchsplanung muss nicht schwierig sein, aber da es sich dabei im Grunde um ein statistisches Thema handelt, liegt es außerhalb des Rahmens dieses Buches. Ich erwähne dieses Thema, weil leider nichts anderes in diesem Buch ein Experiment mit einem schlechten Design retten kann. Vor allem bei Studien mit hohem Durchsatz ist es wichtig, sich Gedanken über die Versuchsplanung zu machen, da technische "Batch-Effekte" die Studien erheblich beeinträchtigen können (siehe dazu Leek et al., 2010).
Die meisten einführenden Statistikkurse und Bücher behandeln grundlegende Themen der Versuchsplanung. Quinn und Keough's Experimental Design and Data Analysis for Biologists (Cambridge University Press, 2002) ist ein hervorragendes Buch zu diesem Thema. Auch in Kapitel 18 von O'Reilly's Statistics in a Nutshell, 2nd Edition, von Sarah Boslaugh werden die Grundlagen gut behandelt. Beachte jedoch, dass die Versuchsplanung in einem Genomik-Experiment eine ganz andere Sache ist und aktiv erforscht und verbessert wird. Der beste Weg, um sicherzustellen, dass dein mehrere tausend Dollar teures Experiment sein Potenzial ausschöpft, ist, sich über die bewährten Methoden für dein Projekt zu informieren. Es ist auch eine gute Idee, deinen befreundeten Statistiker vor Ort zu konsultieren, wenn du Fragen zur Versuchsplanung oder Bedenken hast.
Code für Menschen schreiben, Daten für Computer schreiben
Das Debuggen ist doppelt so schwer wie das Schreiben des Codes. Wenn du also den Code so clever wie möglich schreibst, bist du per Definition nicht clever genug, um ihn zu debuggen.
Brian Kernighan
Bioinformatikprojekte können Berge von Code umfassen, und einer unserer besten Schutzmaßnahmen gegen Fehler ist es, Code für Menschen und nicht für Computer zu schreiben (ein Punkt, der in dem ausgezeichneten Artikel von Wilson et al., 2012). Der Mensch ist es, der die Fehlersuche durchführt, also erleichtert das Schreiben von einfachem, klarem Code die Fehlersuche.
Der Code sollte lesbar sein, in kleine Komponenten aufgeteilt (modular) und wiederverwendbar (damit du den Code nicht immer wieder für dieselben Aufgaben neu schreibst). Diese Praktiken sind in der Softwarewelt von entscheidender Bedeutung und sollten auch bei deiner Arbeit in der Bioinformatik angewendet werden. Das Kommentieren von Code und die Verwendung eines Styleguides sind einfache Methoden, um die Lesbarkeit von Code zu verbessern. Google hatfür viele Sprachen öffentliche Styleguides, die als hervorragende Templates dienen. Warum ist die Lesbarkeit von Code so wichtig? Erstens macht lesbarer Code Projekte besser reproduzierbar, da andere leichter verstehen können, was Skripte tun und wie sie funktionieren. Zweitens ist es viel einfacher, Softwarefehler in lesbarem, gut kommentiertem Code zu finden und zu korrigieren als in unordentlichem Code. Drittens ist es immer einfacher, den Code in der Zukunft zu überarbeiten, wenn er gut kommentiert und klar geschrieben ist. Das Schreiben von modularem und wiederverwendbarem Code erfordert einfach Übung - wir werden im Laufe des Buches einige Beispiele dafür sehen.
Im Gegensatz zu Code sollten Daten auf so formatiert werden, dass sie für den Computer lesbar sind. Allzu oft zeichnen wir Menschen Daten so auf, dass sie für uns möglichst gut lesbar sind, aber erst mit erheblichem Aufwand bereinigt und aufgeräumt werden müssen, bevor sie von einem Computer verarbeitet werden können. Je mehr Daten (und Metadaten) computerlesbar sind, desto besser können wir unsere Computer für die Arbeit mit diesen Daten nutzen.
Lass deinen Computer die Arbeit für dich machen
Menschen, die Tätigkeiten auswendig lernen, neigen dazu, viele Fehler zu machen. Eine der einfachsten Möglichkeiten, deine Arbeit robuster zu machen, besteht darin, so viel wie möglich von dieser Routinearbeit vom Computer erledigen zu lassen. Dieser Ansatz der Automatisierung von Aufgaben ist robuster, weil er die Wahrscheinlichkeit verringert, dass du einen banalen Fehler machst, z. B. versehentlich eine Datei auslässt oder die Ausgabe falsch benennt.
Wenn du zum Beispiel ein Programm auf 20 verschiedene Dateien anwendest, indem du jeden Befehl einzeln eintippst (oder kopierst und einfügst), ist das sehr riskant - die Wahrscheinlichkeit, dass du einen Flüchtigkeitsfehler machst, steigt mit jeder Datei, die du bearbeitest. Bei der Arbeit in der Bioinformatik ist es gut, wenn du dir angewöhnst, diese Art von wiederkehrender Arbeit von deinem Computer erledigen zu lassen. Anstatt 20 Mal denselben Befehl einzufügen und nur die Ein- und Ausgabedateien zu ändern, solltest du ein Skript schreiben, das diese Aufgabe für dich übernimmt. Das ist nicht nur einfacher und weniger fehleranfällig, sondern erhöht auch die Reproduzierbarkeit, da dein Skript als Referenz dafür dient, was du mit jeder dieser Dateien gemacht hast.
Um die Vorteile der Automatisierung von Aufgaben zu nutzen, musst du dir ein paar Gedanken über die Organisation deiner Projekte, Daten und deines Codes machen. Einfache Gewohnheiten wie die einheitliche Benennung von Datendateien, die ein Computer (und nicht nur ein Mensch) verstehen kann, können die Automatisierung von Aufgaben erheblich erleichtern und die Arbeit wesentlich vereinfachen. Beispiele dafür werden wir in Kapitel 2 sehen.
Behauptungen aufstellen und laut sein, im Code und in deinen Methoden
Wenn wir Code schreiben, neigen wir dazu, implizite Annahmen über unsere Daten zu treffen. Wir erwarten zum Beispiel, dass es nur drei DNA-Stränge gibt (vorwärts, rückwärts und unbekannt), dass die Startposition eines Gens kleiner ist als die Endposition und dass es keine negativen Positionen geben kann. Diese impliziten Annahmen, die wir über die Daten machen, beeinflussen die Art und Weise, wie wir unseren Code schreiben; zum Beispiel denken wir vielleicht nicht daran, eine bestimmte Situation im Code zu behandeln, wenn wir davon ausgehen, dass sie nicht auftreten wird. Leider kann dies zu dem gefürchteten stillen Fehler führen: Unser Code oder unsere Programme erhalten Werte, die außerhalb der erwarteten Werte liegen, verhalten sich nicht korrekt und geben trotzdem ohne Warnung eine Ausgabe zurück. Die beste Methode, um diese Art von Fehlern zu vermeiden, ist, unsere Annahmen über Daten in unserem Codeexplizit anzugeben und zu testen, indem wir Assert-Anweisungen wie assert() in Python und stopifnot() in R verwenden.
Fast jede Programmiersprache hat ihre eigene Version der Assert-Funktion. Diese Assert-Funktionen funktionieren auf ähnliche Weise: Wenn die Aussage als falsch ausgewertet wird, hält die Assert-Funktion das Programm an und meldet einen Fehler. Sie mögen einfach sein, aber diese Assert-Funktionen sind für eine robuste Forschung unverzichtbar. Schon früh in meiner Karriere hat mich ein Mentor dazu gebracht, Assert-Funktionen großzügig zu verwenden - auch wenn es so aussieht, als ob die Aussage niemals falsch sein könnte - und ich bin immer wieder überrascht, wie oft sie einen subtilen Fehler entdeckt haben. In der Bioinformatik (und in allen anderen Bereichen) ist es wichtig, dass wir so viel wie möglich tun, um die gefürchteten stillen Fehler in laute Fehler zu verwandeln.
Teste den Code, oder noch besser: Lass den Code den Code testen
Softwareentwickler/innen sind ein schlaues Völkchen, und sie die Idee, den Computer die Arbeit machen zu lassen, auf eine neue Ebene. Eine Möglichkeit, dies zu tun, besteht darin, den Code von anderen zu testen, anstatt ihn von Hand zu machen. Eine gängige Methode zum Testen von Code ist das sogenannte Unit Testing. Beim Unit-Testing zerlegen wir unseren Code in einzelne modulare Einheiten (was auch den Nebeneffekt hat, dass die Lesbarkeit verbessert wird) und schreiben zusätzlichen Code, der diesen Code testet. In der Praxis bedeutet das, wenn wir eine Funktion namens add() haben, schreiben wir eine zusätzliche Funktion (normalerweise in einer separaten Datei) namens test_add(). Diese test_add() Funktion ruft die add()Funktion mit bestimmten Eingaben auf und testet, ob die Ausgabe den Erwartungen entspricht. In Python könnte so aussehen:
EPS=0.00001# a small number to use when comparing floating-point valuesdefadd(x,y):"""Add two things together."""returnx+ydeftest_add():"""Test that the add() function works for a variety of numeric types."""assert(add(2,3)==5)assert(add(-2,3)==1)assert(add(-1,-1)==-2)assert(abs(add(2.4,0.1)-2.5)<EPS)
Die letzte Zeile der Funktion test_add() sieht komplizierter aus als die anderen, weil sie Fließkommazahlen vergleicht. Es ist schwierig, Fließkommawerte auf einem Computer zu vergleichen, da es Darstellungs- und Rundungsfehler gibt. Es ist jedoch eine gute Erinnerung daran, dass wir immer durch die Möglichkeiten unserer Maschine eingeschränkt sind und diese Einschränkungen bei der Analyse berücksichtigen müssen.
Unit-Tests werden in der wissenschaftlichen Programmierung viel weniger eingesetzt als in der Softwareindustrie, obwohl wissenschaftlicher Code eher Fehler enthält (weil unser Code in der Regel nur einmal ausgeführt wird, um Ergebnisse für eine Veröffentlichung zu generieren, und viele Fehler in wissenschaftlichem Code unauffällig sind). Ich bezeichne dies alsdas Paradoxon des wissenschaftlichen Codes: Die Fehleranfälligkeit des wissenschaftlichen Codes bedeutet, dass wir genauso viel oder mehr testen sollten als die Softwareindustrie, aber wir testen tatsächlich viel weniger (wenn überhaupt). Das ist bedauerlich, denn heutzutage sind viele wissenschaftliche Schlussfolgerungen das Ergebnis von Bergen von Code.
Das Testen von Code ist zwar die beste Methode, um Softwarefehler zu finden, zu beheben und zu verhindern, aber das Testen ist nicht billig. Das Testen von Code macht unsere Ergebnisse robuster, aber es kostet uns auch eine Menge Zeit. Leider würde es zu viel Zeit in Anspruch nehmen, wenn Forscher/innen für jedes Stück Code, das sie schreiben, Unit-Tests erstellen würden. Die Wissenschaft entwickelt sich schnell, und in der Zeit, die du für das Schreiben und Ausführen von Unit-Tests benötigst, könnte deine Forschung veraltet sein oder vom Markt verschwinden. Eine sinnvollere Strategie ist es, drei wichtige Variablen zu berücksichtigen, wenn du einen Code schreibst:
-
Wie oft wird dieser Code von anderen Codes aufgerufen?
-
Wenn dieser Code falsch wäre, wie sehr würde sich das auf das Endergebnis auswirken?
-
Wie auffällig wäre ein Fehler, wenn er auftreten würde?
Wie wichtig es ist, einen Teil des Codes zu testen, ist proportional zu den ersten beiden Variablen und umgekehrt proportional zur dritten (d.h. wenn ein Fehler sehr auffällig ist, gibt es weniger Grund, einen Test dafür zu schreiben). Diese Strategie werden wir in allen Beispielen des Buches anwenden.
Bestehende Bibliotheken nutzen, wann immer möglich
Es gibt einen Wendepunkt in der Karriere eines jeden angehenden Programmierers, an dem er sich wohl genug fühlt, um Code zu schreiben und denkt: "Hey, warum sollte ich dafür eine Bibliothek benutzen, das kann ich doch ganz einfach selbst schreiben." Das ist ein ermutigendes Gefühl, aber es gibt gute Gründe, stattdessen eine bestehende Softwarebibliothek zu verwenden.
Bestehende Open-Source-Bibliotheken haben zwei Vorteile gegenüber Bibliotheken, die du selbst schreibst: eine längere Geschichte und ein größeres Publikum. Beide Vorteile führen zu weniger Fehlern. Bugs in Software sind vergleichbar mit dem sprichwörtlichen Problem, eine Nadel im Heuhaufen zu finden. Wenn du deine eigene Softwarebibliothek schreibst (in der zwangsläufig ein paar Bugs lauern), bist du eine Person, die nach ein paar Nadeln sucht. Im Gegensatz dazu haben bei Open-Source-Softwarebibliotheken viel mehr Personen viel länger nach diesen Nadeln gesucht. Daher ist die Wahrscheinlichkeit, dass Fehler in diesen Open-Source-Bibliotheken gefunden, gemeldet und behoben werden, größer als bei deinen eigenen, selbst gebauten Versionen.
Ein gutes Beispiel dafür ist ein potenziell subtiles Problem, das beim Schreiben eines Skripts zur Übersetzung von Nukleotiden in Proteine auftritt. Die meisten Biologen und Biologinnen mit etwas Programmiererfahrung können ein Skript für diese Aufgabe leicht schreiben. Aber hinter diesen einfachen Programmierübungen lauert eine versteckte Komplexität, die du allein vielleicht nicht bedenkst. Was ist, wenn deine Nukleotidsequenzen Ns enthalten? Oder Ys? Oder Ws? Ns, Ys und Ws sehen vielleicht nicht wie gültige Basen aus, aber das sind die Standardnukleotide der International Union of Pure and Applied Chemistry (IUPAC), die in der Bioinformatik durchaus zulässig sind. In vielen Fällen haben gut geprüfte Softwarebibliotheken diese Art von versteckten Problemen bereits gefunden und behoben.
Daten als schreibgeschützt behandeln
Viele Wissenschaftler verbringen viel Zeit mit Excel und ändern ohne mit der Wimper zu zucken den Wert in einer Zelle und speichern die Ergebnisse. Ich rate dringend davon ab, Daten auf diese Weise zu verändern. Stattdessen ist es besser, alle Daten alsschreibgeschützt zu behandeln und Programmen nur zu erlauben, Daten zu lesen und neue, separate Dateien mit Ergebnissen zu erstellen.
Warum ist es in der Bioinformatik wichtig, Daten als schreibgeschützt zu behandeln? Erstens kann das Ändern von Daten an Ort und Stelle leicht zu verfälschten Ergebnissen führen. Nehmen wir zum Beispiel an, du hast ein Skript geschrieben, das eine Datei direkt verändert. Mitten in der Verarbeitung einer großen Datei stößt dein Skript auf einen Fehler und bricht ab. Da du die Originaldatei geändert hast, kannst du die Änderungen nicht rückgängig machen und es noch einmal versuchen (es sei denn, du hast ein Backup)! Die Datei ist also beschädigt und kann nicht mehr verwendet werden.
Zweitens kann man leicht den Überblick darüber verlieren, wie wir eine Datei verändert haben, wenn wir sie an Ort und Stelle ändern. Im Gegensatz zu einem Workflow, bei dem jeder Schritt eine Eingabe- und eine Ausgabedatei hat, gibt eine Datei, die an Ort und Stelle geändert wurde, keinen Hinweis darauf, was wir mit ihr gemacht haben. Wenn wir den Überblick darüber verlieren, wie wir eine Datei geändert haben, und keine Sicherungskopie der Originaldaten haben, sind unsere Änderungen nicht mehr nachvollziehbar.
Daten als schreibgeschützt zu behandeln, mag Wissenschaftlern, die mit der Arbeit in Excel vertraut sind, kontraintuitiv erscheinen, aber es ist für eine solide Forschung unerlässlich (und verhindert Katastrophen und hilft bei der Reproduzierbarkeit). Die anfänglichen Schwierigkeiten sind es wert, denn es schützt die Daten nicht nur vor Beschädigung und falschen Änderungen, sondern fördert auch die Reproduzierbarkeit. Außerdem kann jeder Schritt der Analyse leicht wiederholt werden, da die Eingabedaten vom Programm nicht verändert werden.
Verbringe Zeit damit, häufig verwendete Skripte zu Tools zu entwickeln
Im Laufe deiner Entwicklung als hochqualifizierter Bioinformatiker wirst du einige Skripte erstellen, die du immer wieder verwenden wirst. Dabei kann es sich um Skripte handeln, die Daten aus einer Datenbank herunterladen, einen bestimmten Dateityp verarbeiten oder einfach nur die gleichen hübschen Diagramme erstellen. Diese Skripte können mit anderen Labormitgliedern oder sogar in anderen Laboren verwendet werden. Du solltest dich besonders anstrengen und darauf achten, dass diese häufig genutzten oder gemeinsam genutzten Skripte so robust wie möglich sind. In der Praxis sehe ich diesen Prozess so, dass aus einmaligen Skripten Werkzeuge werden.
Im Gegensatz zu Skripten sind Tools darauf ausgelegt, immer wieder ausgeführt zu werden. Sie sind gut dokumentiert, haben eine explizite Versionierung, verständliche Kommandozeilenargumente und werden in einem gemeinsamen Repository mit Versionskontrolle aufbewahrt. Diese Unterschiede mögen unbedeutend erscheinen, aber bei robuster Forschung geht es darum, kleine Dinge zu tun, die dir helfen, Fehler zu vermeiden. Skripte, die du immer wieder auf zahlreiche Datensätze anwendest, wirken sich naturgemäß auf mehr Ergebnisse aus und sollten daher besser entwickelt werden, damit sie robuster und benutzerfreundlicher sind. Das gilt vor allem für Skripte, die du mit anderen Forschern teilst, die die Dokumentation einsehen und dein Tool sicher auf ihre eigenen Daten anwenden können müssen. Die Entwicklung von Tools ist zwar arbeitsintensiver als das Schreiben eines einzelnen Skripts, aber auf lange Sicht spart es Zeit und vermeidet Kopfschmerzen.
Lass die Daten beweisen, dass sie von hoher Qualität sind
Wenn Wissenschaftlerinnen und Wissenschaftler an die Analyse von Daten denken, denken sie in der Regel an die Analyse von experimentellen Daten, um biologische Schlussfolgerungen zu ziehen. Um eine solide bioinformatische Arbeit zu leisten, müssen wir jedoch mehr als nur experimentelle Daten analysieren. Dazu gehört auch die Überprüfung und Analyse von Daten über die Datenqualität deines Experiments, Zwischenausgabedateien von Bioinformatikprogrammen und möglicherweise simulierte Testdaten. Auf diese Weise stellen wir sicher, dass unsere Datenverarbeitung so funktioniert, wie wir es erwarten, und verkörpern die goldene Regel der Bioinformatik: Vertraue weder deinen Tools noch deinen Daten.
Es ist wichtig, dass du nie davon ausgehst, dass ein Datensatz von hoher Qualität ist. Vielmehr sollte die Qualität der Daten durch eine explorative Datenanalyse (auch EDA genannt) überprüft werden. EDA ist weder kompliziert noch zeitaufwändig und macht deine Forschung viel robuster gegenüber Überraschungen, die in großen Datensätzen lauern. In Kapitel 8 erfährst du mehr über EDA mit R.
Empfehlungen für reproduzierbare Forschung
Die Einführung reproduzierbarer Forschungsmethoden erfordert nicht viel zusätzlichen Aufwand. Und genau wie robuste Forschungspraktiken machen reproduzierbare Methoden dein Leben einfacher, da du deine Arbeit vielleicht auch dann noch reproduzieren musst, wenn du die Details schon längst vergessen hast. Im Folgenden findest du einige grundlegende Empfehlungen, die du bei der Bioinformatik beachten solltest, um deine Arbeit reproduzierbar zu machen.
Gib deinen Code und deine Daten frei
Das absolute Minimum für die Reproduzierbarkeit ist, dass Code und Daten veröffentlicht werden. Ohne verfügbaren Code und Daten ist deine Forschung nicht reproduzierbar (siehe Peng, 2001 für eine gute Diskussion darüber). Wie du Code und Daten weitergibst, besprechen wir etwas später in diesem Buch.
Alles dokumentieren
Wenn ein Wissenschaftler zum ersten Mal ein Labor betritt, wird ihm gesagt, dass er ein Labornotizbuch führen soll. Leider wird diese gute Praxis von Forschern in der Informatik oft vernachlässigt. Die Veröffentlichung von Code und Daten ist die Mindestanforderung für Reproduzierbarkeit, aber auch eine ausführliche Dokumentation ist ein wichtiger Bestandteil der Reproduzierbarkeit. Um eine Studie vollständig zu reproduzieren, muss jeder Analyseschritt viel detaillierter beschrieben werden, als es in einem wissenschaftlichen Artikel möglich ist. Daher ist eine zusätzliche Dokumentation für die Reproduzierbarkeit unerlässlich.
Eine gute Praxis ist es, jeden deiner Analyseschritte in README-Dateien im Klartext zu dokumentieren. Wie ein detailliertes Notizbuch dient diese Dokumentation als wertvolle Aufzeichnung deiner Schritte, wo sich Dateien befinden, woher sie kommen oder was sie enthalten. Diese Dokumentation kann zusammen mit dem Code und den Daten deines Projekts aufbewahrt werden (mehr dazu in den Kapiteln 2 und 5), damit deine Mitstreiter herausfinden können, was du getan hast. Die Dokumentation sollte auch alle Eingabeparameter für jedes ausgeführte Programm, die Versionen dieser Programme und die Art und Weise, wie sie ausgeführt wurden, enthalten. Moderne Software wie R's knitr und iPython Notebooks sind leistungsstarke Werkzeuge für die Dokumentation von Forschungsergebnissen; ich habe in der README dieses Kapitels auf GitHub einige Ressourcen für den Einstieg in diese Werkzeuge aufgelistet.
Mach Zahlen und Statistiken zu Ergebnissen von Skripten
Um sicherzustellen, dass ein wissenschaftliches Projekt reproduzierbar ist, geht es nicht nur um die Reproduzierbarkeit der wichtigsten statistischen Tests, die für die Ergebnisse wichtig sind, sondern auch um die Reproduzierbarkeit der unterstützenden Elemente einer Arbeit (z. B. Abbildungen und Tabellen). Der beste Weg, um sicherzustellen, dass diese Komponenten reproduzierbar sind, ist, dass jedes Bild oder jede Tabelle die Ausgabe eines Skripts (oder mehrerer Skripte) ist.
Das Schreiben von Skripten zur Erstellung von Bildern und Tabellen mag zeitaufwändiger erscheinen als die interaktive Generierung in Excel oder R. Wenn du jedoch schon einmal mehrere Zahlen von Hand neu generieren musstest, nachdem du einen früheren Schritt geändert hast, kennst du die Vorzüge dieses Ansatzes. Skripte, die Tabellen und Bilder erzeugen, lassen sich leicht wiederholen, sparen Zeit und machen deine Forschung reproduzierbarer. Tools wie iPython Notebooks und knitr (im vorigen Abschnitt erwähnt) sind auch bei diesen Aufgaben sehr hilfreich.
Code als Dokumentation verwenden
Bei komplexen Verarbeitungspipelines ist die beste Dokumentation oft ein gut dokumentierter Code. Da der Code ausreicht, um einem Computer mitzuteilen, wie er ein Programm ausführen soll (und welche Parameter er verwenden soll), reicht er auch fast aus, um einem Menschen zu sagen, wie er deine Arbeit reproduzieren kann (zusätzliche Informationen wie die Softwareversion und die Eingabedaten sind ebenfalls notwendig, um vollständig reproduzierbar zu sein). In vielen Fällen ist es einfacher, ein Skript zu schreiben, um die wichtigsten Schritte einer Analyse auszuführen, als Befehle einzugeben und sie dann an anderer Stelle zu dokumentieren. Auch hier gilt: Code ist eine wunderbare Sache. Wenn du jeden Analyseschritt mit Code dokumentierst, ist es einfach, alle Schritte einer Analyse zu wiederholen, wenn es nötig ist - das Skript kann einfach erneut ausgeführt werden.
Verbessere deine Bioinformatik-Datenkenntnisse kontinuierlich
Behalte die in diesem Kapitel vorgestellten Grundgedanken im Hinterkopf, wenn du den Rest des Buches durcharbeitest. Was ich hier vorgestellt habe, reicht gerade aus, um dich mit einigen Kernkonzepten der robusten und reproduzierbaren Bioinformatik vertraut zu machen. An vielen dieser Themen (z. B. Reproduzierbarkeit und Softwaretests) wird derzeit noch aktiv geforscht, und ich möchte interessierte Leser dazu ermutigen, sich eingehender damit zu befassen (ich habe einige Ressourcen in der README dieses Kapitels auf GitHub bereitgestellt).
Get Bioinformatik Datenkenntnisse now with the O’Reilly learning platform.
O’Reilly members experience books, live events, courses curated by job role, and more from O’Reilly and nearly 200 top publishers.

